Peutz-Jeghers Syndrome (PJS) is a rare genetic disorder with its own characteristics that deserve adequate knowledge and individualized management.
It is transmitted in an autosomal dominant manner in affected families, but it can also appear sporadically (i.e. without family history). Autosomal means that it is not linked to sex, i.e. it is in the chromosomes that are the same in men and women; dominant means that only one copy of the gene (of 2 existing) that is defective is enough for the disease to be expressed. A mutation has been detected in the STK11 gene, which would be the basis of several (but not all) cases of this syndrome.
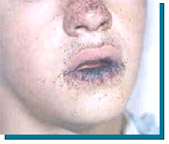
The main characteristics of this syndrome are the presence of melanic spots on the skin and mucous membranes (see photo), which generally appear in the first years of life and tend to disappear in adulthood, and hamartomatous polyps in the gastrointestinal tract. These two elements can occur to a greater or lesser degree in different individuals and in different members of the same family.
The most frequent reason for consultation is abdominal pain (“cramps”), a consequence of the presence of polyps in the intestine. They can also cause digestive bleeding and anemia. There is an increased risk of suffering from intestinal or extraintestinal cancer, so lifelong monitoring is required (there are “monitoring guidelines” for this). These guidelines are not strict in themselves, the important thing is to have regular check-ups with the doctor.
The risk of a child of an individual with this syndrome having the disease is 50%. Treatment must be appropriate for each patient.
There is a downloadable SPJ information book available (see below) which is intended for children, but adults can find many answers and important information in it.